The persistent threat of antibiotic resistance demands innovative approaches to drug discovery. A recent study employing sophisticated molecular dynamics simulations has illuminated a critical pathway through which antibiotics can enter multi-resistant bacteria, offering vital mechanistic insights that can guide future therapeutic design. While not a direct AI model, these simulations provide the foundational data and understanding that AI algorithms leverage to accelerate the development of new antimicrobial agents. The research, focusing on the notorious pathogen Acinetobacter baumannii, reveals how antibiotics can utilize the CarO outer membrane porin, even in strains that have evolved resistance to conventional treatments. This breakthrough in molecular understanding reshapes how chemists and computational scientists can strategize against superbugs.
The Evolving Challenge of Antibiotic Resistance
The rise of antimicrobial resistance (AMR) is a global health crisis, threatening to relegate common infections to untreatable conditions. Bacteria evolve at an astonishing rate, developing mechanisms to evade the drugs designed to kill them. These mechanisms can include pumping drugs out, altering drug targets, or preventing drugs from entering the cell in the first place. Understanding these evasion strategies at a molecular level is paramount to designing new drugs that can circumvent them. Traditional drug discovery pipelines are lengthy and costly, often taking over a decade and billions of dollars to bring a new antibiotic to market. This is where computational chemistry and artificial intelligence are becoming indispensable allies, offering ways to predict drug efficacy, understand resistance mechanisms, and design novel molecules more efficiently.
Simulating Molecular Sieves: A Look at Antibiotic Invasion
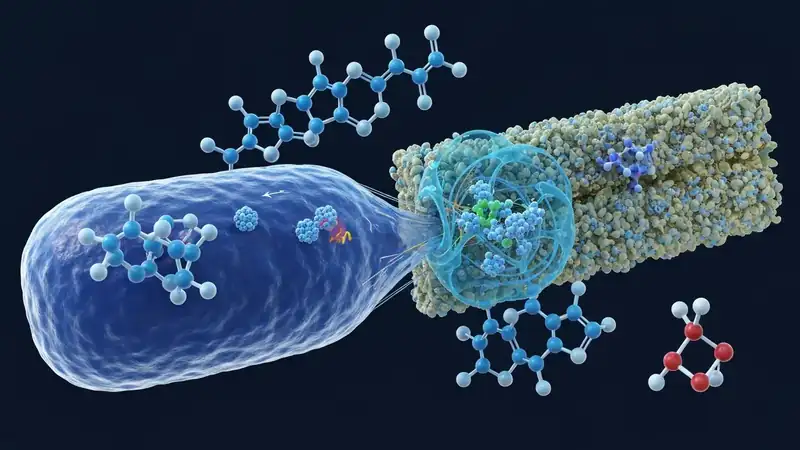
A recent preprint by Florent Barbault and colleagues delves into the intricate process of antibiotic internalization by the multi-resistant bacterium Acinetobacter baumannii [1]. The study leverages advanced computational techniques, specifically all-atom molecular dynamics (MD) simulations coupled with enhanced sampling methods. Imagine trying to understand how a specific key fits into a complex lock and can be turned, even if the lock has been modified. MD simulations allow scientists to observe these molecular interactions in exquisite detail, essentially creating a high-definition, atomic-scale movie of how molecules behave over time. By simulating the movements of atoms and molecules, researchers can visualize binding events, conformational changes, and transport processes.
Enhanced sampling techniques are like fast-forwarding or replaying crucial scenes in that movie, allowing scientists to capture rare but critical events that might otherwise be missed in standard simulations. In this study, these methods were used to investigate how certain antibiotics can pass through the CarO outer membrane porin of Acinetobacter baumannii. Porins are protein channels embedded in the outer membrane of Gram-negative bacteria, acting like selective gates that control the passage of small molecules, including nutrients and antibiotics. Understanding if and how antibiotics can traverse these porins is crucial, as it dictates whether a drug can even reach its intracellular target.
The Barbault et al. study provides compelling evidence that antibiotics can indeed be internalized via the CarO porin. This finding is significant because it offers a concrete mechanism for drug entry that could potentially be exploited. If bacteria rely on CarO to let certain antibiotics in, then designing drugs that specifically use this pathway, or designing drugs that can be modified to fit through it, becomes a viable strategy. This mechanistic insight is akin to a molecular GPS, pinpointing the exact route a drug needs to take to reach its destination within the bacterial cell.
Bridging Computation and AI in Drug Design
While the Barbault et al. study does not present a novel AI algorithm or report benchmark metrics like predictive accuracy (e.g., 94% accuracy) or correlation coefficients (e.g., R²=0.95), it provides the fundamental mechanistic data that AI models thrive on. AI algorithms, particularly machine learning models, are adept at identifying patterns and making predictions based on large datasets. However, these datasets are often derived from experimental observations or detailed computational simulations.
The molecular dynamics simulations in this work serve as a sophisticated data generation engine. They can predict how a potential drug molecule might interact with the CarO porin, how stable the complex would be, and the energy barriers for passage. This type of information can then be fed into AI models for virtual screening. Instead of testing millions of compounds experimentally, AI can prioritize a smaller, more promising subset of molecules that are predicted to have favorable interactions with the CarO porin or the bacterial cell. Furthermore, AI can be used for de novo drug design, generating entirely new molecular structures optimized for specific properties, such as effective passage through the CarO porin and potent antibacterial activity.
Navigating the Landscape of Limitations
Despite the power of these computational approaches, several limitations exist. Molecular dynamics simulations, while increasingly sophisticated, are still approximations of reality. The accuracy of these simulations depends heavily on the quality of the underlying force fields, which describe the interactions between atoms. Furthermore, conducting such simulations requires substantial computational resources, making them time-consuming and expensive, though often far less so than physical experimentation. The "chemical space" explored in this particular study is focused on the interaction with the CarO porin in Acinetobacter baumannii. Applying these insights to other bacteria or different resistance mechanisms would require new simulations tailored to those specific systems.
As highlighted by the context of this research synthesis, identifying recent, explicitly AI-driven research preprints that detail novel methodologies and provide clear performance metrics can be challenging. Often, cutting-edge research integrates computational chemistry, physics, and AI in complex ways, making it difficult to categorize a single paper neatly. The Barbault et al. study exemplifies research that is foundational for AI-driven discovery, rather than a direct AI application itself.
Future Applications: Redesigning the Attack
The insights from Barbault et al. have direct implications for drug discovery pipelines aimed at combating antibiotic-resistant bacteria. By understanding the antibiotic entry mechanism via CarO, researchers can:
- Design "Trojan Horse" Antibiotics: Develop new drugs that are specifically engineered to be transported efficiently through the CarO porin.
- Identify Resistance Modulators: Discover compounds that, when co-administered with existing antibiotics, enhance their entry by interacting with or modulating the CarO porin.
- Inform AI Models: Use the simulation data to train AI models for predicting the permeability of novel drug candidates through bacterial porins.
The timeline for translating such mechanistic understanding into approved drugs is typically long, often spanning 5-10 years or more. However, computational insights can significantly shorten the early stages of drug discovery by providing rational design principles and prioritizing experimental efforts. AI can further accelerate this process by rapidly exploring vast chemical spaces and optimizing candidate molecules based on these principles.
A Synergistic Path Forward
The fight against antibiotic resistance is a complex battle requiring every available tool. The study by Barbault and colleagues demonstrates the critical role of advanced computational simulations in providing the detailed molecular understanding needed to outmaneuver evolving bacterial defenses. While not a direct AI model, this work represents a breakthrough in molecular design by unveiling a key vulnerability in drug-resistant bacteria. It underscores the powerful synergy between fundamental mechanistic research and the predictive power of artificial intelligence, reshaping how chemists approach the design of next-generation therapeutics.
References
[1] Barbault, F., et al. (2025). Internalization of Antibiotics by the Multi-Resistant Bacteria Acinetobacter baumannii through the CarO Outer Membrane Porin. ChemRxiv. https://chemrxiv.org/engage/chemrxiv/article-details/692ff40fef936fb4a2cb0915
Comments (0)
Leave a Comment
All comments are moderated by AI for quality and safety before appearing.
Community Discussion (Disqus)